Zhang Wei Saimei Feier Technology
Tan Qingqiao Zhongxin Guojian Pharmaceutical
1. Foreword
Glycosylation modification is one of the most extensive and important post-translational modifications of life activities, affecting not only the spatial conformation, biological activity, transport and localization of proteins, but also specific organisms such as molecular recognition, cell communication, and signal transduction. The process plays a vital role. Glycoprotein mainly has two types of N-glycoprotein and O-glycoprotein according to its sugar chain structure and glycosylation site. It is inferred that more than 50% of proteins have glycosylation modifications, but due to the high complexity of glycosylation, most glycoproteins have not been discovered, and only about 10% of proteins in existing databases are annotated as sugar. Protein [1]. First, the composition and structure of the sugar chain is very complicated. The sugar chain is non-template synthesized and has a two-dimensional structure. The glycosidic bond position and conformation are different to form a fine structure. According to reports, the structure of oligosaccharides consisting of only 6 different monosaccharides may reach an astonishing 1012 species [2]. Secondly, when a sugar chain with a composition and a structure is linked to form a glycoprotein on a protein, it also constitutes a microscopic heterogeneity, that is, a problem of diversification of a sugar chain structure at a glycosylation site, and a protein may have multiple glycosyl groups. Sites, and there may be multiple structural sugar chains at each site [3].
With the development of glycoprotein drugs represented by monoclonal antibodies, mass spectrometry has been widely used in glycosylation analysis. Usually, the sugar chains are released from glycoproteins by enzyme digestion or chemical methods, and then the glycosylation sites are identified. And the analysis of the structure of the sugar chain [4]. However, there is no mature technology for the analysis of intact glycopeptides. In this paper, we use the HCD/ETD “double fragmentation mode†of the new generation combined mass spectrometer LTQ-Orbitrap Elite to analyze the complete glycopeptide of a complex glycoprotein. Using the latest Byonic and SimGlycan software analysis, we successfully found two N-glycosyl groups. The chemistry site, 16 O-glycosylation sites, 83 site-specific sugar chain composition and structural information, the protein sequence coverage was as high as 92.1% in the undeglysed state.
2. Experimental part
2.1 Sample Information
The sample is a highly expressed glycosylated protein. The pretreatment uses dithiothreitol (DTT), iodoacetic acid (IAA) to reductive alkylation of disulfide bonds, trypsin to digest protein, and final concentration. It is 1.5 μg/μL.
2.2 Liquid chromatography method
Column: nanoflow rate C18 (2 μm, 100 A, 75 μm x 50 cm), normal flow rate HILIC-NH2 (3 μm, 100 A, 2.0 mm x 15 cm)
Chromatograph: C18 column uses nano-flow liquid phase Easy-nLC 1000, HILIC-NH2 column uses conventional flow rate liquid phase Accela 600 Load: 2 μL
Mobile phase: A: 0.1% aqueous formic acid, B: 0.1% formic acid acetonitrile solution
Gradient: C18: 3%-90% B, 120 min, 250 nL/min; HILIC-NH2: 95%-5% B, 120 min, 200 μL/min
2.3 mass spectrometry
Mass Spectrometer: LTQ-Orbitrap Elite Combined High Resolution Mass Spectrometer
source of ion:
Easy-nLC 1000 uses EASY-Spray nano-current spray ion source: spray voltage 2.3 kV, transfer capillary temperature: 275 ° C, S-Lens: 60%;
Accela 600 uses HESI-II to heat electrospray ion source: spray voltage 3.5 kV,
Sheath gas: 30, auxiliary gas: 10, transfer capillary temperature: 275 ° C, SLens: 60%
Scanning method: data-dependent scanning: one-level full scan (resolution 60,000), Top15 two-level full scan (resolution 15,000); scanning range: parent ion m/z 400-3500, daughter ion m/z 100-3500; Dynamic exclusion: time 30s, exclusion range m/z -0.5~+1.5
Fragmentation mode: high energy collision induced dissociation HCD (NCE 35%), electron transfer dissociation ETD (100 ms for 2+)
2.4 Data Analysis
Data were analyzed using the Byonic software library to identify glycopeptides/non-glycopeptides. The specific parameters were: enzyme: trypsin (KR); specificity: full enzyme digestion; missed cleavage site: 2; parent ion mass accuracy: 15 ppm; Mass accuracy: 20 mD; fixed modification: C+58.005; variable modification: M+15.995; glycosylation modification: N, O; peptide card value standard: Score>150, Delta Mod>5, Log Probability<-1 Glycosylation site card value standard: Score>190, Delta Mod>10, Log Probability<-2. The SEQUEST search library parameters are the same, and the card value standard is 5% FDR. The glycopeptide sugar chain moiety structure was resolved using SimGlycan software.
3. Results and discussion
3.1 Analysis Strategy
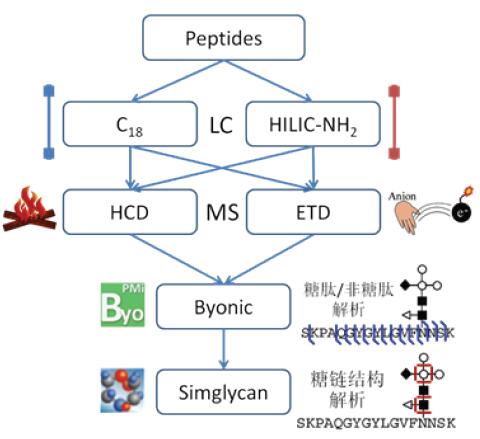
The overall strategy of the experiment is shown in Figure 1. In this experiment, samples were chromatographed using nano-C18 and amino HILIC separation methods. C18 is a commonly used protein/peptide separation column, and the C18 column at a nanoflow rate provides a significant increase in the unit concentration of the analyte, and the sensitivity is optimized to efficiently analyze peptides with low abundance/low ionization efficiency. Amino HILIC is a hydrophilic chromatogram, which is separated according to oligosaccharide/glycan composition and structure. It is an effective tool for sugar chain structure analysis and glycomics research.
At the same time, because the glycosylation of the target protein is highly complex, this experiment uses both HCD and ETD fragmentation modes for mass spectrometry. HCD tends to fragment low-charge/low-molecular peptides. When glycosylation is modified, preferential glycosidic linkages between monosaccharides are broken, while high-energy fragmentation can obtain more fragmentation information. ETD tends to fragment high-charge/high-molecular peptides, preferentially fragmenting the amide bond of the peptide backbone in the presence of glycosylation. Therefore, HCD and ETD have good complementarity, and the combination of the two can be effectively applied to the analysis of intact glycopeptides.
Figure 1. Glycopeptide analysis flow chart
The glycopeptide spectrum contains both the peptide skeleton and the fragment information of the sugar chain, so the analysis of the mass spectrometry data is also an analysis difficulty. Byonic is a professional glycopeptide analysis software that uses an innovative "Preview" algorithm and a rich database of sugar chains to identify peptides and glycosylation sites, and to provide sugar chain composition based on exact mass. SimGlycan is a professional sugar chain structure analysis software with theoretical fragmentation information of 9,650 sugar chains. It is currently the largest commercial sugar database. In this experiment, Byonic was used to sequence the data to obtain the glycosylation site and sugar chain monosaccharide composition information, and then use SimGlycan to analyze the data of the sugar chain structure, and finally obtain the glycopeptide sequence, glycosylation site and position. Point-specific sugar chain structure.
Figure 2. Sample LC-MS/MS Total Ion Flow Diagram (TIC)
3.2 Sequence identification
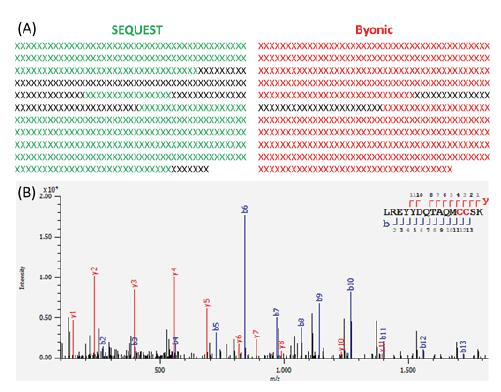
Figure 2 shows the TIC image of the sample obtained by the nano-C18+HCD process analysis. The chromatographic peaks are evenly distributed to achieve the desired separation effect, and the mass spectrometry response reaches 9th power. All mass spectral data were sequenced by Byonic, and the protein sequence coverage was 92.1%. The traditional algorithm SEQUEST was used for searching, and the sequence coverage was only 76.6% (Fig. 3).
Figure 3. (A) SEQUEST/Byonic sequence identification coverage comparison; (B) Byonic pair peptide secondary spectrum matching
The SEQUEST algorithm was unable to identify intact glycopeptides and only based on peptide analysis sequences that did not undergo glycosylation, some peptides that were fully glycosylated were difficult to resolve. In contrast, Byonic identification, based on its sugar chain database, glycosylation as a variable modification, thus enabling direct resolution of glycopeptides. Therefore, Byonic's analytical glycoprotein sequence coverage is significantly improved.
3.3 glycopeptide analysis
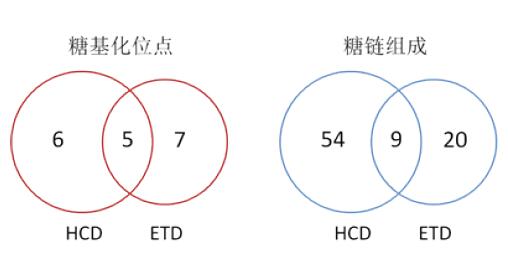
In the above Byonic identification results, a total of 28 glycopeptides were included, corresponding to 18 glycosylation sites, of which 2 N-glycosylation sites and 16 O-glycosylation sites; The point-specific sugar chains are composed of 83 species, corresponding to a maximum of 17 species per site, a minimum of 1 species, and an average of 5.2 species per site. The two fragmentation modes of HCD and ETD show excellent complementarity, resulting in a significant increase in analytical results (Figure 4).
Figure 4. Comparison of glycopeptide analysis results obtained by fragmentation of HCD/ETD
By comparing the mass spectrum of TKPREEQYNSTYR N-glycopeptide (N317) (Fig. 5), it can be seen that both ETD and HCD successfully identified this glycopeptide, and the fragment information (c/z) obtained by ETD is more abundant. The site is covered by c and z ions at the same time. In contrast, the fragmentation information (b/y) obtained by HCD was significantly reduced and the sites were not covered by fragment ions. However, it is worth noting that there are significant cluster peaks at the high molecular weight end of the HCD spectrum, and the peaks differ by 162 Da (Δm/z 81, 2+) or 203 Da (Δm/z 101.5, 2+), indicating the sugar of the glycopeptide. Fragmentation of the chain portion.
Figure 5. HCD/ETD fragmentation spectrum analysis of glycopeptide TKPREEQYNSTYR
ETD tends to fragment the amide bond between the peptide backbone amino acids and retain the modifying group. Therefore, the glycopeptide completely retains the sugar chain in the ETD, and obtains sufficient peptide skeleton fragments (c/z series). More efficient identification of peptides. HCD tends to fragment the weaker post-translational modification group, causing the glycochain of the glycopeptide to be significantly fragmented in HCD mode, presenting a series of fragment peaks separated by a monosaccharide, and the fragmentation of the peptide backbone is significantly inhibited. The b/y series ion is insufficient and the signal is weak; at the same time, the fragment with the complete sugar chain is difficult to generate, so the b/y fragment information including the site N cannot be obtained.
For O-glycopeptides with smaller sugar chains, the effect of HCD on sugar chains is not as large as that of N-glycopeptides, and the signal response of ETD fragmentation (103) is significantly weaker than HCD (104), so in general, two The number of glycopeptides and sites identified was comparable, demonstrating excellent complementarity.
Analysis of 3.4 site-specific sugar chain structure
Byonic does not consider the information of the sugar chain fragments, and only obtains the composition information of the sugar chain based on the exact molecular weight of the peptide fragments. This experiment further uses SimGlycan to analyze the specific structure of the sugar chain. SimGlycan interprets the sugar chain structure based on the information of the sugar chain fragments. Therefore, while analyzing the specific structure, the identification results of Byonic are verified on the other hand, and the final result is more credible.
Figure 6. HCD fragmentation map of glycopeptide TKPREEQYNSTYR
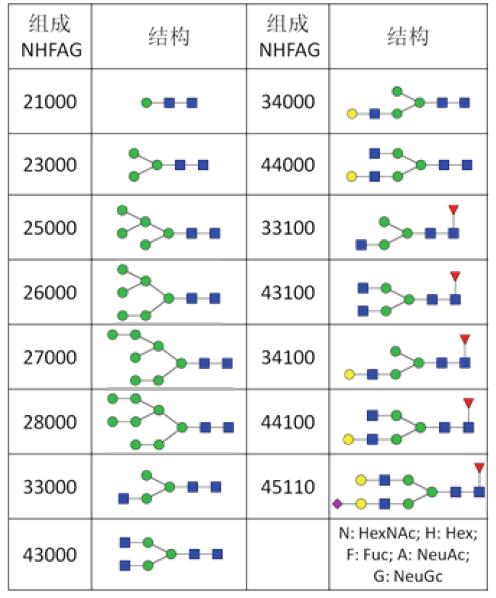
In this experiment, SimGlycan analysis was performed on all the N317 glycosylation sites, and the final 75.6% of the spectra were verified. The corresponding 93.8% of the sugar chains obtained structural information. As shown in Table 1, there are 15 kinds of sugar chain structures in the N317 locus, mainly high-mannose type and double-stranded complex N-glycan chain, and also contain important structures such as core fucose and sialic acid. Figure 6 further shows the results of the analysis of the sugar chain structure of the HCD spectrum of Figure 5 and a partial fragment matching map. The sufficient information of the sugar chain fragment ions indicates that the structure of Hex2HexNAc8 is a three-branched high mannose type.
Analysis of 3.4 site-specific sugar chain structure
Byonic does not consider the information of the sugar chain fragments, and only obtains the composition information of the sugar chain based on the exact molecular weight of the peptide fragments. This experiment further uses SimGlycan to analyze the specific structure of the sugar chain. SimGlycan interprets the sugar chain structure based on the information of the sugar chain fragments. Therefore, while analyzing the specific structure, the identification results of Byonic are verified on the other hand, and the final result is more credible.
Table 1. Sugar chain structure of N317 site
In addition, there are many isomers in the sugar chain, especially the fine structure differences with the same composition and different connection modes, which are difficult to distinguish only by the secondary fragments. Of course, the multi-level resolution capability (MSn) of LTQ-Orbitrap Elite also enables efficient fine-grained analysis of sugar chains.
4. Conclusion
This paper uses a new generation of combined mass spectrometry LTQ-Orbitrap Elite to successfully analyze the integrity of a highly glycosylated protein using the leading HCD/ETD "double fragmentation mode" combined with advanced glycosylation analysis software Byonic and Simglycan. Glycopeptide. In the experiment, two N-glycosylation sites, 16 O-glycosylation sites, and 83 site-specific sugar chain compositions and structures were obtained. The protein sequence coverage was as high as 92.1% in the undeglyzed state. The efficient analysis of complex glycoproteins is realized. The technology-leading LTQ-Orbitrap Elite is a powerful tool for complex macromolecular analysis in the fields of glycomics, proteomics and biopharmaceuticals.
references:
[1] Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database [J]. Biochim. Biophys. Acta 1999, 1473 (1): 4-8.
[2] Morelle, W.; Canis, K.; Chirat, F.; Faid, V.; Michalski, JC The use of mass spectrometry for the proteomic analysis of glycosylation [J]. Proteomics 2006, 6 (14): 3993 -4015.
[3] Dai Jingquan, Cai Wei, Qian Xiaohong; Protein glycosylation analysis method and its application in proteomics[J]. Biotechnology Communication 2005, 16 (3): 287-292.
[4] Geyer, H.; Geyer, R. Strategies for analysis of glycoprotein glycosylation [J]. Biochim. Biophys. Acta 2006, 1764 (12): 1853-1869.
Foot Spa Massager
Foot Spa Massager,Bath Foot Massager Machine,Foot Spa Bath,Bath Foot Massager With Bubble
Huaian Mimir Electric Appliance Co., LTD , https://www.mimirfootbath.com